An intuitive look at the xcorr score in proteomics
in Proteomics
Proteomics에서 사용되는 xcorr score에 대한 직관적 이해 [작성중]
출처: https://willfondrie.com/2019/02/an-intuitive-look-at-the-xcorr-score-function-in-proteomics/
Peptide identification을 위한 database search는 기본적으로 theoretical spectrum과 measured/observed spectrum을 비교하는 작업이다. 두 spectrum을 비교하기 위해서는 적절한 similarity measure가 필요한데 가장 대표적인 database search engine인 SEQUEST는 xcorr을 사용한다. 그렇다면 xcorr는 어떤 의미를 갖는 것일까?
xcorr를 이해하기 위해서는 먼저 cross-correlation을 이해해야 한다. Cross-correlation은 신호처리에서 많이 사용되는 metric인데 시간의 함수로 표현되는 두 signal간의 similarity를 나타낸다.
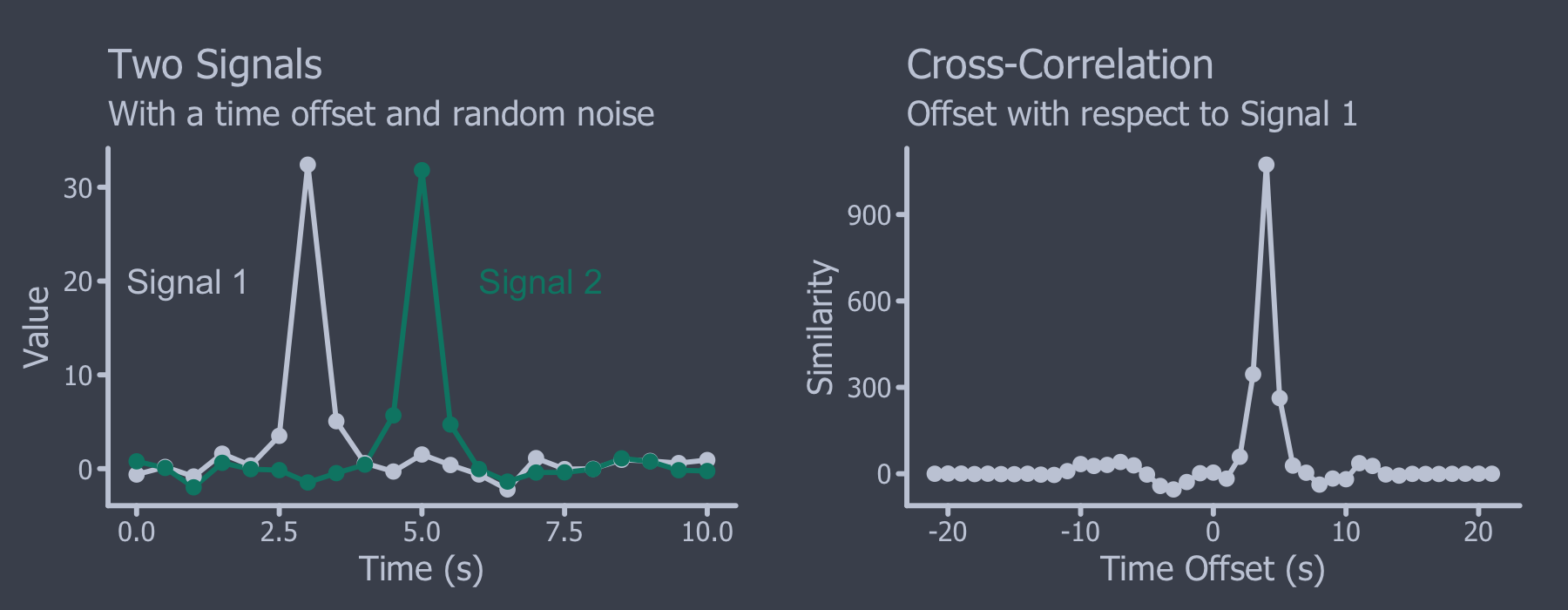
위의 그림과 같이 offset이 있는 두 신호의 경우에 offset이 align된 경우에는 높은 cross-correlation값을 가지며, 그렇지 않은 경우에는 낮은 값을 갖게 된다. Cross-correlation은 각각의 offset에서 두 신호의 dot product와 동일하므로, sliding dot product라고도 불린다.
Mass spectrometry의 경우는 m/z domain에서 정의되는 신호로서 time대신에 m/z offset을 고려하게 되는데, 두 신호가 m/z offset이 없으면 높은 cross-correlation값을 그렇지 않을 경우에는 낮은 값을 갖게 된다. SEQUEST에서 사용되는 xcorr는 다음과 같이 정의된다.
[수식]
즉, m/z offset = 0일때의 cross-correlation과 그렇지 않을 때의 cross-correlation사이의 차이를 보는 것이다.